By Allan Muir
With contributions from George Fox and Jared Salbato
Friday, November 17, 2006
Texas Hoe Down at Sea World
Report by George Fox
 On Friday evening after check in and registration, the AMDA hosted a reception “Hoe Down” at near by Sea World. The entire group had access to some of Sea World’s amenities and we had the park all to ourselves. The first attraction was the Dolphin tank where we were able to feed and interact with the dolphins. There were about 8-10 dolphins in a huge swimming pool like tank that allowed us to get close enough to feed by hand. These dolphins were very smart and powerful creatures and it was great to have such closer interaction. By the time we were finished I’m sure the dolphins had plenty of energy and full bellies to boot. It seems they would have kept eating as long as we kept feeding. It was the first time I have ever seen a dolphin with a double chin.
On Friday evening after check in and registration, the AMDA hosted a reception “Hoe Down” at near by Sea World. The entire group had access to some of Sea World’s amenities and we had the park all to ourselves. The first attraction was the Dolphin tank where we were able to feed and interact with the dolphins. There were about 8-10 dolphins in a huge swimming pool like tank that allowed us to get close enough to feed by hand. These dolphins were very smart and powerful creatures and it was great to have such closer interaction. By the time we were finished I’m sure the dolphins had plenty of energy and full bellies to boot. It seems they would have kept eating as long as we kept feeding. It was the first time I have ever seen a dolphin with a double chin.
 Next stop was the main aquarium. Just outside the main aquarium, to our surprise, was an appetizer buffet as well as a real live country western band. I believe it was the Lost Mule Band. Whatever their name was, they were a very talented, 5 piece band that carried an authentic country western sound and had lots of good tunes. I could have listened all night, however there were a few sharks that wanted to have dinner with us in the main aquarium.
Next stop was the main aquarium. Just outside the main aquarium, to our surprise, was an appetizer buffet as well as a real live country western band. I believe it was the Lost Mule Band. Whatever their name was, they were a very talented, 5 piece band that carried an authentic country western sound and had lots of good tunes. I could have listened all night, however there were a few sharks that wanted to have dinner with us in the main aquarium.
Walking into the main entrance of this underwater seascape was like walking into a big cave lined with smaller aquariums that housed a multitude of underwater sea creatures. This tunnel led to a huge banquet sized room that had curved walls that were actually the glass sides of the main aquarium. The aquarium was home to several different species of fish and marine life. Most notable to me were the Sand Tiger sharks that were as big as pick-up trucks. The good news was, there was a large piece of glass between us and the sharks that allowed us to make funny faces at them without fear of loosing an arm. The other good news was that this great room was where we were to be treated to an authentic western Barbecue with ribs, corn on the cob, roast beef, pecan pie and a host of other favourites. It was a dining room anyone would be envious of.
 After dinner the Lost Mules were still laying down some classic square dancing favorites and there was a group of folks cutting a rug to a number of different songs. The song I remember most was the “Bull $hi+” song. This song uses a move normally reserved for getting dung off of ones boot. This kicking motion was incorporated into a line dance that captured everyone’s attention; very clever! After a great night we loaded every one back onto the busses, wheel chairs were no problem, and headed back to the hotel for a night cap and a good night sleep.
After dinner the Lost Mules were still laying down some classic square dancing favorites and there was a group of folks cutting a rug to a number of different songs. The song I remember most was the “Bull $hi+” song. This song uses a move normally reserved for getting dung off of ones boot. This kicking motion was incorporated into a line dance that captured everyone’s attention; very clever! After a great night we loaded every one back onto the busses, wheel chairs were no problem, and headed back to the hotel for a night cap and a good night sleep.
Saturday, November 18, 2006
Opening Remarks
 Randall House, IPA Chairman and AMDA President
Randall House, IPA Chairman and AMDA President
Randall opened the conference by declaring that 2006 had been a “Tremendous year for the Pompe Community”, and talked of the broad label market approval of Myozyme by both EMEA and the FDA. He looked back over the last 12 to 15 years commenting on the major and rapid advancements in understanding the disease and its treatment since his daughter Tiffany was diagnosed some 13 years ago. At the time of her diagnosis there was no patient organisation they could turn to and there was a very loose-knit scientific community investigating the disease. Randall was justly proud to say that in the early days of the AMDA they were able to pull together those individuals and form invaluable collaborations. It was a difficult, long and expensive road that has culminated in Myozyme. He added that Myozyme is the first of many steps towards finding enhanced treatments. Randal gave thanks to those researchers and to Genzyme that we now, al last, have a therapy for Pompe Disease.
 Arnold Reuser, PhD, Master of Ceremonies
Arnold Reuser, PhD, Master of Ceremonies
Randall handed over to Arnold Reuser who was to act as Master of Ceremonies throughout the conference. In fact Arnold exceeded his duties by giving a brief and amusing presentation of what we should expect from the conference, as well as taking responsibilities for the hall lighting, and fixing the numerous computer and sound system problems over the weekend.
Panel A
Pompe Disease: Clinical and Patient Perspectives
Introduction to Panel A
Y T Chen, M.D.
 YT Chen reiterated Randall’s statement that “this is just the beginning” before handing over to the first speaker in this panel. He introduced Priya Kishnani reminding us that she had been working with Pompe patients well before the first AMDA conference and that she was working in the lab before clinical trials were started.
YT Chen reiterated Randall’s statement that “this is just the beginning” before handing over to the first speaker in this panel. He introduced Priya Kishnani reminding us that she had been working with Pompe patients well before the first AMDA conference and that she was working in the lab before clinical trials were started.
Updates on Pompe Disease – Management and Treatment
Priya Kishnani
Priya opened by stating that the broad label for Pompe disease was an historic event because it was so unusual for a treatment for adults to be approved on the basis of trials involving only infants. Usually it would be the other way around.
Priya gave an overview of the infantile trials AGLU1602 and AGLU1702 and explained how they had given a better understanding of CRIM infants, i.e. those who produce no measurable GAA. These infants were more likely to develop antibodies, including some that inhibited myozyme and all CRIM children had responded less well to Myozyme. She said that there was a need for intervention to suppress the immune response which may lead to the possibility of using lower
doses of the therapy.
Moving on to the need for a multidisciplinary approach to caring for Pompe children Priya discussed the recent “Pompe Disease Diagnosis and Management Guidelines” published by the American College of Medical Genetics. These are available at: www.acmg.net/resources/policies/Pompe_Disease.pdf. The guidelines include recommendations for the following areas:
- Diagnostics
- Cardiology
- Pulmonary
- Gastrointestinal/Nutritional
- Musculoskeletal/Functional/Rehabilitation
- Neurology
- General Medical Care
- Surgery Anaesthesia
The guidelines will also hopefully help families fight against insurance companies who are reluctant to help. Priya said that all trial subjects had shown positive changes to therapy but that the motor function component was the limiting factor and stressed the need for appropriate physiotherapy. She also pointed out that an accumulation of brain fluid prior to treatment had resolved in 4 out of 6 patients and was resolving in the remaining 2. This is something requiring further monitoring. Priya was very concerned about hearing loss in many of the children; out of 22 subjects 12 suffered fluid collection in their ears, she noted however that the middle-ear fluid does reduce with time.
In conclusion Priya said that there was still a lot to understand about the treated infant, to understand the long-tem motor response that is so diverse across all patients. She contrasted the poor response of some infants with the case of a 60 year old patient showing a remarkable improvement after 6 months of therapy. She said that there were interesting times ahead with the hope of newer therapies to improve the enzyme targeting, enzyme chaperone treatments and advances in gene therapy.
Long Term effects of ERT in Pompe Disease
Ans Van Der Ploeg
Ans began by showing the long line of research at the Erasmus Medical Centre, Rotterdam, which led to the eventual proof of concept of Enzyme Replacement Therapy for Pompe disease. Of the current team she highlighted the 1977 PhD thesis of Arnold Reuser and her own in 1989. Clinical trials in Rotterdam for Myozyme started in 2003 and Marloes Hagemans PhD Thesis was published this year on the Natural Course of Pompe Disease.
Ans stressed how the cooperation between industry, patient organisations and universities had been so important to the development of a treatment for Pompe disease.
Discussing lessons learnt from the trials she said that the results so far showed the importance of early treatment. Patients showing initial improvement can often decline, often due to respiratory problems. She illustrated this with pictures of one little girl suffering almost complete paralysis with little chance of improvement. Ans said that Myozyme had brought significant improvements but the balance is delicate; treated children are always prone to develop further symptoms of the disease.
For late-onset patients Ans said that the Mini-LOTS trial (Juveniles) had shown significant improvement in lung function. In some, muscle strength had revived but in others it had not improved, only stabilised. The LOTS trial is ongoing at Rotterdam but results will not be made public until 2007.
Muscle Pathology was discussed; Ans said that patients can lose 40% of their muscle tissue before a severe loss of strength becomes evident. Muscle wasting in infants is much more rapid in infants than late-onset patients. Disease progression in infants can take between 3 to 7 months whilst the same wasting might take 10 or 30 years in an adult. Where a patient maintains good muscle condition ERT may be able to improve up to 75% of the muscle fibres.
Muscle atrophy can occur for several reasons in the Pompe patient: Immobilisation, starvation, the aging process, infectious disease or for many other reasons e.g. cancer. Ans advised patients to maintain a healthy diet and to engage in moderate (sub-maximal) exercise. For advice on diet and exercise she referred to a recent Muscle & Nerve article by Dr Alf Slonim; “Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy”.
The Role of Patient Organisations
Ria Broekgaarden
Ria gave a very comprehensive summary of the history, work, challenges and achievements of the International Pompe Association, a federation of around 35 patient groups around the globe. She began by stating the mission and objectives of the association which can be viewed on the IPA website, www.worldpompe.org.
In the early days of clinical trials the IPA was instrumental in helping Pharming and Genzyme find suitable patients. This was due to those companies limited exposure to neuromuscular physicians; most of their contacts from studies of other diseases had been with metabolic centres.
Ria discussed the diverse nature of patient groups around the world, ranging from single points of contact to organised groups such as the AMDA. She discussed the formation of the IPA and how the collaboration across countries and with industry and academia had led to several notable achievements. She cited the Erasmus MC Questionnaire and the Pompe Connections brochures as great achievements we should be particularly proud of.
She discussed some of the past challenges that had to be overcome: Misleading information (for example statements in the past that therapy would be available by the year 2000) and the animal rights lobby. She talked of difficult times during the transition from different production methods, particularly transferring patients from the Transgenic (rabbit milk) enzyme to the synthetic (CHO) product.
Through the IPA, patients were able to give the patient perspective at hearings held by EMEA and the FDA. These were instrumental in gaining a broad label for Myozyme and in the future similar presentations will hopefully facilitate reimbursement for patients where treatment is denied.
The “Panel A” Q&A session involving Priya, Ans and Ria raised some interesting points:

Priya, Ans and Ria
- The level of naturally produced enzyme does not drive the response to therapy.
- Criteria for starting treatment: For infants this should be the first weeks of life. For adults we need to know at what stage the disease is reversible. If no symptoms appear then perhaps it is too early but the recommendation was that if a patient has a chance of therapy, don’t wait.
- The ethical question of treating CRIM was discussed given that they have such poor outcome. It seemed that there was still much uncertainty about the nature of CRIM status although many families are keen to know the status of their babies.
- The majority of adults have the same mutation and yet the age of disease onset can be very different.
- Osteoporosis seems to be a common feature of neuromuscular conditions.
Panel B
Pompe Disease: Corporate and Governmental Perspectives
Arnold Reuser introduced the second panel.
 Genzyme: Corporate Perspective
Genzyme: Corporate Perspective
Frank Ollington
Frank gave a personal reflection of his involvement whilst leading the Pompe project at Genzyme. He used three words to summarise his impression: Urgency, Complexity and Global.
He recalled times when European ministers called the CEO demanding access to the drug in response to parents who were chaining themselves to the railings outside government buildings.
But he also talked of the achievement of the collective effort of physicians, scientists, industry, patients and their advocates, combining to form a community. He said that Genzyme had been inspired by patients for over 25 years but never more so than by the Pompe patients. He was honoured and privileged to work on their behalf and found them inspirational.
He assured the audience that Genzyme will continue to spend $100s over the coming years to improve therapy for Pompe disease and to market the drug effectively. Their commitment is ongoing. “It is not an option to stay still,” he said.
 Genzyme: Corporate Perspective
Genzyme: Corporate Perspective
Paul Merrigan
Paul is the Vice President for the Global Marketing of Pompe Disease. He started by saying that the emphasis at Genzyme had progressed from the “urgency for a treatment” to the “urgency to treat”, pointing out that Myozyme already has market approval in 27 countries world-wide. He added that a further 20 countries are currently considering approval. Reimbursement is available in a total of 22 countries and overall there are patients on treatment in 37 countries. Paul announced that just this week (ending 17 Nov 2006) Genzyme’s target for 2006 of 500 patients on treatment has been reached.
Paul talked about how Genzyme were working to improve the pathway to early diagnosis and treatment. He also discussed health economics and the burden of disease and noted that there are currently 264 patients enrolled onto the Genzyme Pompe Registry.
 Genzyme Treatment Support
Genzyme Treatment Support
Sara Dundon and Kathleen Brewer
 Sara and Kathleen are part of the Genzyme Treatment Support team who advise patients on issues such as legislation and insurance. Their advice is free, confidential and voluntary. They act to educate the patient, physician and funder on the disease awareness and its treatment. They can also take responsibility for coordination of care for an individual.
Sara and Kathleen are part of the Genzyme Treatment Support team who advise patients on issues such as legislation and insurance. Their advice is free, confidential and voluntary. They act to educate the patient, physician and funder on the disease awareness and its treatment. They can also take responsibility for coordination of care for an individual.
The danger of insurance limitations was discussed. For example some policies have life-time or annual maximum payments. There are various ways in which Patient Services Incorporated (PSI) can help when these maxima are exceeded. Suggestions included access to the state “High Risk Pool” run by some 30 states. Help in converting existing insurance policies can be given and in some instances funds can be accessed though a patients professional, alumni or occupational associations.
 Food and Drug Administration
Food and Drug Administration
JoAnn Minor
JoAnn’s presentation tried to answer the questions “who does what and when?” and “is the FDA too cautious or not cautious enough?”
For the three parties – FDA, drug developer, and patient = JoAnn gave an illustration of the process of reaching drug approval from R&D stages and through the initial IND (investigational New Drug) application lodged by the drug developer. This process can take typically 13 years and $800M from the discovery of a new molecule to the development of a new drug. JoAnn illustrated the drug development timeline with the diagram below:
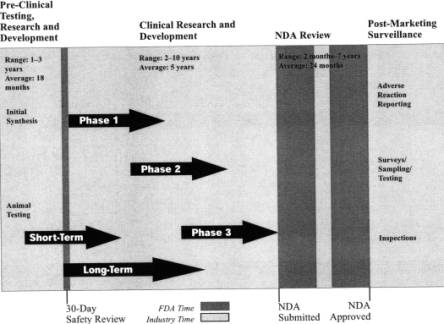
New Drug Development Timeline
Panel B Q&A made the following points:
- The LOTS study will hopefully be completed in March 2007 however it maybe extended to improve statistical confidence in the data. This is the last opportunity to run such a study as all patients gain access to therapy so it is crucial that the highest quality of data is obtained. Results from the study will be submitted at a major medical meeting for review and it is hoped that a report will be available by 3rd quarter of 2007.
- One patient who started the trial in November 2005 was disappointed to hear of the trial extension and might wish to be transferred to the commercial product.
- Where adults are being denied funding from their insurance companies it was suggested that trying the “juvenile-onset” approach had worked for some.
- There was no consensus on the benefits of diet or supplements (L-Alanine).
 Presentation of Pompe Connections, Treatment Edition
Presentation of Pompe Connections, Treatment Edition
Maryze Schoneveld van der Linde
Maryze announced that the “Treatment Edition” of the Pompe connections has now been published. This a comprehensive set of brochures on Myozyme infusions, management, cost and expected outcomes. It is an IPA publication with contributions from professionals and patients, but most of the credit must go to Maryze for her fine efforts in coordinating the publication. This edition is accessible from the IPA website but currently only available in English. The other leaflets in the series are available in English, Dutch, Turkish, German, Spanish, and French. They can be viewed or downloaded from the IPA website, www.worldpompe.org. The Pompe Connections Treatment Edition will also be translated in several languages so that as many people as possible can access the information.
 Panel C
Panel C
Living with Pompe Disease
Rochelle Hirshhorn introduced Panel C
 Speech and Swallowing Issues in the Neurologic Population
Speech and Swallowing Issues in the Neurologic Population
Sharon Veis
Sharon gave a detailed account of the many aspects of speech and communication; she discussed both their acquisition during childhood and their degeneration as a result of disease. The mechanism of speech was described and with the aid of slides and videos she showed the effects of several disorders.
Sharon highlighted the need for more published information and noted that Duke University is soon to hold a meeting on the subject. Without this information speech therapists can find treating Pompe patients very challenging.
For Pompe disease an evaluation of all aspects of speech are necessary: the musculature of the lips, tongue, palate, and larynx. Also the respiratory muscles and any breathing support all play a part.
Sharon fully described the mechanisms of swallowing and showed how it should be monitored for both efficiency and safety (in terms of risk of choking). To illustrate this we were shown X-ray videos of patients exhibiting problems relevant to Pompe disease.
 Prevention of Respiratory Complications of Pompe Disease
Prevention of Respiratory Complications of Pompe Disease
Dr John Bach
Dr. Bach began his presentation by saying how upset he was to see Pompe patients with tracheotomies. It is his belief that non-invasive ventilation (NIV) was appropriate in all cases. He later added that trached patients become ventilator dependant and become prone to infections and pneumonias.
During his talk he showed patients suffering with Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig’s disease, a progressive neuromuscular disease. These patients had no measurable lung-function but he managed them successfully with NIV.
He blamed the problem on pulmonists who are only used to dealing with lung and airway disease, not muscle disease. The most common errors in the care of patients with neuromuscular disease were:
- Misinterpretation of symptoms
- Inadequate pulmonary function tests
- Failure to monitor sleep properly (“sleep studies are a waste of time”)
- Over reliance on measurements of Arterial Blood Gases (ABGs).
- Over reliance on tracheotomies
- Over reliance on suctioning
- Giving oxygen therapy
Many patients have a normal ability to breathe, but no ability to cough which requires 4 times greater volume. This must be treated effectively (e.g. by a cough-assist Mechanical Insufflation-Exsufflation device.) and the ability to cough should be monitored using a peak-flow meter.
He advocated volumetric ventilators rather than pressure devices (such as the BiPap) as they ensure that the lungs are fully inflated and have no positive pressure during exhalation – a feature developed to prevent sleep apnoea but not usually necessary for neuromuscular disease.
At one point during the presentation he had the whole audience practicing “Frog Breathing”. This is a method of gulping air into the lungs without using the normal respiratory muscles. At the time I thought I was successfully breathing in this way, but afterwards I have had little success. It obviously takes a lot of practice but could be very useful in an emergency.
John’s final comment was that every Pompe patient should have an oximeter in the house. He then referred us to his numerous books, papers and his website: www.doctorbach.com.
 Therapeutic Intervention Guidelines for Patients with Infantile and Late-Onset Pompe Disease
Therapeutic Intervention Guidelines for Patients with Infantile and Late-Onset Pompe Disease
Dawn Phillips
The goals of these guidelines were to enable patients to improve their sitting comfort and quality, to improve their mobility, and to enable a standing transfer. Improved mobility in patients has been shown to increase their special awareness and also helps reduce the occurrence of contractures. Improving special awareness has been shown to lead to improved cognitive abilities. Dawn showed how young patients can be supported with equipment to help them stand and move around their environment.
Dawn discussed strategies for helping late-onset patients whom she classed as Severe, Sitters, and Ambulators. She discussed the merits of articulating Ankle-Foot Orthoses (AFO) for preventing contractures; it is believed that stretching muscles and tendons beyond their at-rest length for at least 6 hours per day is beneficial.
Discussing exercise, Dawn recommended sub-maximal exercise for 45 minutes at a time, two or three times per week. During exercise the heart should reach a maximum rate given by the equation:
Heart rate = (220 – age) x 0.65 beats per minute.
To summarise, the table below shows the maximum recommended heart rate in beats per minute as a function of age.
| Age (years) |
Maximum Heart Rate (Beats per minute) |
|---|---|
|
10 |
137 |
|
20 |
130 |
|
30 |
124 |
|
40 |
117 |
|
50 |
111 |
|
60 |
104 |
|
70 |
98 |
Panel C
Q&A Points from the Panel C questions included:
- A BiPap is no good for getting a deep breath and the exhalation pressure is too high.
- Heavy weights should not be used to rebuild muscle strength. Low resistance is recommended initially with increasing repetitions rather than weight.
Sunday—November 19, 2006
Panel C Continued on Sunday:
 Natural Course of Pompe Disease
Natural Course of Pompe Disease
Marloes Hagemans
Marloes based her presentation on analysis of data derived from the IPA/Erasmus MC patient survey. She reported that she now had collected baseline questionnaires from 314 Pompe patients around the globe and that 3-year follow-up questionnaires had been sent to 189 participants. Her most recent analysis concerned patients sharing the same genetic mutations but presenting a different disease course. Looking at genetic differences she found that 68% were actually the same despite diagnoses occurring at anywhere between 1 and 30 years of age. For this IVS1 mutation the residual enzyme could vary anywhere between 4 and 20%. So it is clear that secondary factors must explain the disease onset and progression. These factors could be related to nutrition, exercise, growth, muscle fibre type, or glycogen metabolism for example.
Marloes reported that the 2-year follow-up results had surprisingly shown significant disease progression for the late-onset Pompe population. The data measured the disease impact on daily life and work. It may appear obvious to individuals in the Pompe world, but it is very important to be able to measure the group response. Fatigue was found to be more prevalent than previously thought, even in those mildly affected.
Marloes concluded that the survey had led to a much better understanding of the disease. This is of great relevance to the treatment decisions and the results are complimentary to all the lab measurements made during patient evaluations.
The results have fed into the FDA guidance for treatment of patients. They will help identify prognostic factors and the timing of ERT. They will also be used to evaluate the long-tem affects of therapies.

Panel D
The Future of Pompe Disease Research
Rodney Howell, M.D., introduced the “Pompe Disease Research” presentations
Muscle Pathology and Mechanisms of Muscle Wasting
Nina Raben MD, PhD
Report by George Fox
 Nina Raben, a brilliant research scientist from the National Institutes of Health (NIH), the largest research facility in the world, had a presentation that described some of the findings her group has had in relation to muscle pathology in Pompe disease. The most striking finding they have had is a slowly evolving shift in the belief that excessive lysosomal glycogen storage is the only problem in Pompe. In fact, her research shows that glycogen filled lysosomes may be a secondary problem to the more serious autophagic build-up. It is these autophagic clusters that seem to have a clogging effect on the cells degradation system. This “biological garbage” is not able to be removed because of defective lysosomal operation. Autophagy, as it is called, is a normal cellular process that gets described as the “house cleaning” system of a cell. Lysosomes, endosomes, and autophagosomes are all involved in keeping the muscle cells free of biological waste. This complicated re-cycling system is rendered defective in Pompe due to the lack of appropriate amount of GAA in lysosomes. The glycogen that accumulates in these lysosomes makes them ineffective and therefore cannot complete there job. If the lysosomes cannot do their job the “trash” that accumulates can be toxic.
Nina Raben, a brilliant research scientist from the National Institutes of Health (NIH), the largest research facility in the world, had a presentation that described some of the findings her group has had in relation to muscle pathology in Pompe disease. The most striking finding they have had is a slowly evolving shift in the belief that excessive lysosomal glycogen storage is the only problem in Pompe. In fact, her research shows that glycogen filled lysosomes may be a secondary problem to the more serious autophagic build-up. It is these autophagic clusters that seem to have a clogging effect on the cells degradation system. This “biological garbage” is not able to be removed because of defective lysosomal operation. Autophagy, as it is called, is a normal cellular process that gets described as the “house cleaning” system of a cell. Lysosomes, endosomes, and autophagosomes are all involved in keeping the muscle cells free of biological waste. This complicated re-cycling system is rendered defective in Pompe due to the lack of appropriate amount of GAA in lysosomes. The glycogen that accumulates in these lysosomes makes them ineffective and therefore cannot complete there job. If the lysosomes cannot do their job the “trash” that accumulates can be toxic.
It is important to note that this build up is primarily found in type II or fast twitch muscle fibres. The other important point to make is that this autophagic build-up has implications for ERT, since the enzyme seems to be mistargeted in cells with autophagic build-up. The enzyme doesn’t make it to the lysosome where it can degrade the glycogen and may explain why the type II fibres don’t respond as well as Type I to ERT.
I like to compare this process to the garbage trucks that come around once a week to pick up our household trash. The trucks would be the lysosomes. If you took your trash out to the side of the street every week and all the garbage truck did was come by and wave and tell you he was full, eventually there would be a huge pile of garbage outside your house. Not only your trash, but all your neighbours’ trash and so on, would accumulate and eventually you would have trash all down the middle of the street. This accumulated trash would stop the flow of traffic and eventually the whole neighbourhood would shut down. So the problem starts with the full garbage truck or lysosomes. However, the toxicity and insult and reason for failure is the accumulated trash. So, we need to figure out how to get the glycogen out of the lysosomes so they can do their job OR find an alternate means of removing the trash that accumulates. Nina’s presentation was top notch and state of the art. There were very detailed confocal microscopic images and three-dimensional colour enhanced images that I have never seen before that were so stunning. Images that allowed us to peer down the core of intact muscle cells whose centres were lined with autophagic build up. A very impressive presentation, we are extremely lucky to have Nina and the NIH on our side.
Pompe Disease, the quest for other Contributing Factors
Robert Mattaliano, PhD
Report by Jared Salbato
 Bob Mattaliano is Vice President of Protein Development for Genzyme Corp. He gave a very interesting presentation on secondary factors which may explain some of the variance seen in patients with respect to disease presentation as well as response to therapy. His talk consisted mainly of genetic factors although there are likely environmental factors as well. It is quite clear that the severity of the mutations in the GAA gene don’t tell the entire story. A wonderful example of this is the IVS1 mutation. This mutation is found in ~80% of adults with Pompe Disease. It is considered a “mild” mutation since some normal, fully active enzyme, is produced from this “blueprint” although at reduced levels. An example of the variance seen in patients with this mutation can be seen in two patients with very different presentations. The first, a four-year old with severe disease and the second, a 30 year old, who is virtually asymptomatic. Clearly, there are other contribution factors. Other genes may be acting as severity modifiers; some forms of these genes may have a protective effect and some may worsen the severity. Bob and his team chose a handful of candidate genes which were hand chosen (my words) as genes that are likely to act as disease modifiers based on the current models of Pompe disease.
Bob Mattaliano is Vice President of Protein Development for Genzyme Corp. He gave a very interesting presentation on secondary factors which may explain some of the variance seen in patients with respect to disease presentation as well as response to therapy. His talk consisted mainly of genetic factors although there are likely environmental factors as well. It is quite clear that the severity of the mutations in the GAA gene don’t tell the entire story. A wonderful example of this is the IVS1 mutation. This mutation is found in ~80% of adults with Pompe Disease. It is considered a “mild” mutation since some normal, fully active enzyme, is produced from this “blueprint” although at reduced levels. An example of the variance seen in patients with this mutation can be seen in two patients with very different presentations. The first, a four-year old with severe disease and the second, a 30 year old, who is virtually asymptomatic. Clearly, there are other contribution factors. Other genes may be acting as severity modifiers; some forms of these genes may have a protective effect and some may worsen the severity. Bob and his team chose a handful of candidate genes which were hand chosen (my words) as genes that are likely to act as disease modifiers based on the current models of Pompe disease.
To date greater than 300 mutations have been identified in the GAA gene. A large percentage represents pvt mutations, meaning they have so far only been found in individual patients. Patients who share some of the more common mutation have varying levels of residual enzyme activity as well as disease severity. To further understand this, Bob’s team looked at haplotypes of the gene in patients containing the IVS1 mutation. If I understood correctly, he was looking for other non-disease causing changes in the gene which are inherited in conjunction with the disease causing IVS1 mutation. These are called polymorphisms. They are non-disease causing variants. These changes did not explain the variance either. Lastly, Bob’s group looked at gene expression analysis from muscle biopsies from patients to look for changes in the levels of gene products which may explain why some patients respond well to therapy and some do not.
By using “gene chips” researchers can simultaneously look at thousands of genes and using complex statistical analysis determine which genes show a statistically significant change in gene expression levels. Bob showed a list of 20-30 genes which were identified using this method.
Bob and his team at Genzyme continue to provide valuable research into Pompe disease. We are all grateful for their contributions.
The Next Generation and Gene Therapeutics for Pompe Disease
Seng Cheng, PhD
Report by Jared Salbato
 Seng Cheng gave a very interesting presentation on “Next Generation Enzyme and Gene Therapeutics for Pompe Disease.” Seng Cheng is Vice President of Genetic Disease Science at Genzyme Corp. He is working on an improved version of Myozyme which he calls neo-rhGAA which shows improved delivery to skeletal muscle compared to Myozyme. The clearance of glycogen from the skeletal muscle requires high dosages of enzyme. Currently, it is not completely clear why this is the case. One reason may be that the affinity of Myozyme for the mannose-6-phosphate receptor is low. Seng Cheng and his team have added additional mannose-6-phosphate residues to the enzyme in order to increase the enzyme’s affinity towards the Mannose-6-phosphate receptor. While Myozyme only contains 1 mannose-6-phosphate on average, neo-rhGAA contains 15-17 mannose-6-phosphate residues. Neo-rhGAA shows improved delivery to skeletal muscle and glycogen clearance and results in a 5-10 fold increased potency. Seng Cheng showed side-by-side comparisons between Myozyme, neo-rhGAA, and saline controls in terms of enzyme activity in skeletal muscle, glycogen clearance, and rotarod performance of Pompe mice. A rotarod is a spinning rod, and the length of time that the mice can walk on this rod is a measure of skeletal muscle function. Neo-rhGAA showed improved efficacy versus Myozyme and Saline control for all parameters tested.
Seng Cheng gave a very interesting presentation on “Next Generation Enzyme and Gene Therapeutics for Pompe Disease.” Seng Cheng is Vice President of Genetic Disease Science at Genzyme Corp. He is working on an improved version of Myozyme which he calls neo-rhGAA which shows improved delivery to skeletal muscle compared to Myozyme. The clearance of glycogen from the skeletal muscle requires high dosages of enzyme. Currently, it is not completely clear why this is the case. One reason may be that the affinity of Myozyme for the mannose-6-phosphate receptor is low. Seng Cheng and his team have added additional mannose-6-phosphate residues to the enzyme in order to increase the enzyme’s affinity towards the Mannose-6-phosphate receptor. While Myozyme only contains 1 mannose-6-phosphate on average, neo-rhGAA contains 15-17 mannose-6-phosphate residues. Neo-rhGAA shows improved delivery to skeletal muscle and glycogen clearance and results in a 5-10 fold increased potency. Seng Cheng showed side-by-side comparisons between Myozyme, neo-rhGAA, and saline controls in terms of enzyme activity in skeletal muscle, glycogen clearance, and rotarod performance of Pompe mice. A rotarod is a spinning rod, and the length of time that the mice can walk on this rod is a measure of skeletal muscle function. Neo-rhGAA showed improved efficacy versus Myozyme and Saline control for all parameters tested.
He also showed that the skeletal muscle of older Pompe mice were more resistant to enzyme treatment. The reasons are not clear but it may be related to the decrease in Mannose-6-Phosphate receptors with age, extent of muscle damage, or the depletion of satellite cells (cells involved in muscle repair) with age. Lastly, Seng Cheng showed data on gene therapy in Pompe mice. He showed the effects of gene therapy versus ERT using either Myozyme or neo-rhGAA. The response to treatment seemed to be significantly more robust than either forms of ERT in terms of glycogen clearance and rotorod performance. The more robust response is likely due to the increased exposure of the mice to enzyme since gene therapy results in high level, continuous production of enzyme by the liver.
 Newborn Screening for the Lysosomal Storage Disorders
Newborn Screening for the Lysosomal Storage Disorders
Joan Keutzer, PhD
Joan described the enzyme assay using bloodspots taken from newborn babies several days after birth. The technique was based on work by Néstor A. Chamoles, M.D. from Argentina who sadly died in May 2005. The method improved by Genzyme could be available by late 2007 and has been shown to yield far fewer false positives than Newborn Screening (NBS) analyses for other diseases.
Referring to the US decision process, Joan said that there are strict criteria for including diseases in national newborn screening programs: There must be a therapy available, there should be clear benefits afforded by the therapy as shown by clinical studies, and there must be an inexpensive test available. Joan showed how all these criteria were now almost satisfied and it is hoped that the screening will be adopted by the US and a number of other countries in the future.
To drive the message home Joan compared the development of six Taiwanese infants; three were diagnosed by NBS and the other three diagnosed conventionally. The three identified by NBS had normal muscle strength and two had cardiomegaly, they were each treated with Myozyme before they were one month old. The three infants treated later are performing significantly less well.
Panel D Q&A
Questions during this session formed the following points:
- Type I and Type II muscle fibres represent red and white meat.
- Glycogen in the diaphragm clears beautifully in mice. It is not known how human diaphragm responds as a muscle biopsy would be required.
- Timelines for new therapies: Still developing the ligand for neoGAA, the decision on a way forward will be made in 2007. Gene therapy has a lot yet to learn from other trials (Parkinson’s, haemophilia). Researchers are tracking and learning from those studies.
- Duchene Muscular Dystrophy trials are currently underway.
- Report in “Nature” reported that a Duchene MD dog treated with stem cells A recent responded remarkably well. Stem cell research is currently “half a step” behind gene therapy.
- There is extreme heterogeneity in muscle fibres. Some fibres exhibit severe glycogen storage whilst their immediate neighbours might be unaffected.
- It is not known if mice maintain their gains in muscle strength over the long term.
- The liver is responsible for degrading the infused enzyme; neoGAA does not improve this liver-sink response.
Closing of the Conference
Randall House, IPA Chairman and AMDA President
Randal closed the conference after thanking all those involved for making it a highly successful occasion.
Panel E
Group Discussion
Chaired by Maryze Schoneveld van der Linde (IPA) and Brian White

After the conference had formerly closed a patient meeting was convened to allow patients and families discuss their views openly and hear the experiences of others. Amongst the deliberations some of the points raised were:
Benefits of treatment
- Taking whole sentences
- Fewer afternoon sleeps
- Small but significant changes
- Not so breathless
- Fewer headaches
Reactions to treatment
- One report of retching, feeling pale, clammy, flushing
Exercise during infusion
- Maryze exercises for 1 hour during her infusion. No evidence that it helps but it seems logical to get the blood pumping into the muscles.
Osteoporosis
- Several patients reported very low bone density and frequent fractures.
- One reported that his son has very good bone density.
Alcohol
- Alcohol induces breathing difficulties
- Reduces stamina and strength
- May affect sleep.
- Makes you feel warmer
Other support
- Take up singing or a wind-instrument to improve strength of respiratory muscles.
GSDNet
- GSDNet holds a huge amount of personal experience. Someone should produce a database to enable further analysis.
Patients need to educate physicians and nursing staff to enable the optimum level of care.
- Problems with IV infusions. To aid the insertion of the IV needle:
- Good hydration is important – drink plenty beforehand
- Keep warm.

(adapted from the AMDA website: amda-pompe.org/2006-conference/)